Purpose in short: I want to detect (and remove) potential contaminants in the genome assembly, and Kraken is a tool designed for that.
Introduction
From its webpage:
Kraken is a system for assigning taxonomic labels to short DNA sequences, usually obtained through metagenomic studies. Previous attempts by other bioinformatics software to accomplish this task have often used sequence alignment or machine learning techniques that were quite slow, leading to the development of less sensitive but much faster abundance estimation programs. Kraken aims to achieve high sensitivity and high speed by utilizing exact alignments of k-mers and a novel classification algorithm.
In its fastest mode of operation, for a simulated metagenome of 100 bp reads, Kraken processed over 4 million reads per minute on a single core, over 900 times faster than Megablast and over 11 times faster than the abundance estimation program MetaPhlAn. Kraken’s accuracy is comparable with Megablast, with slightly lower sensitivity and very high precision.
Kraken is written in C++ and Perl, and is designed for use with the Linux operating system. We have also successfully compiled and run it under the Mac OS.
In practice
See Kraken manual for full instructions.
Install
Download the latest release.
1 | unzip kraken-1.1.zip && cd kraken-1.1 |
If you want to build your own dabase, jellyfish version 1 should be in your PATH.
Build standard Kraken database
The standard Kraken database includes bacterial, archeal, and viral genomes in Refseq at the time of the build.
1 | !/bin/sh |
If any step (including the initial downloads) fails, the build process will abort. However, kraken-build will produce checkpoints throughout the installation process, and will restart the build at the last incomplete step if you attempt to run the same command again on a partially-built database.
Build custom Kraken database
Because the standard Kraken database doesen’t include fungi and protozoa, which I want to include in my analysis.
I found refseq2kraken, which facilitates the downloading and preparation of Kraken database.
1 | path2kraken=$TOOLDIR/kraken.1.1 |
In the following part of this note, this library will be refered as to ‘Non-Masked library’.
This post Download refseq-genomic data and prepare it for Kraken is also helpful.
Mask low-complexity regions
I noticed a pull request of kraken: Fixed human genome downloading and added auto-masking feature using dustmasker, and realized that I should mask the library! So I re-generated the kraken library (I don’t know why the author of kraken didn’t mention that in their docs.)
Here is the new script:
1 | !/bin/sh |
In the following part, this library will be refered as to ‘Masked library’.
Classify contigs
Non-Masked library
1 | classify |
The ‘classified’ sequences will be saved in ‘classified.fa’, and the ‘unclassified.fa’ will be the ‘clean’ one, which can be used for downstream analysis.
But when I looked over the ‘report’, the result quite disappointed me.
1 | head -5 flye.run2.report |
This means about 70% contigs were contaminated!
I was curious about the percentage of contaminant lengths of each read. Then I used a simple Python script to count that.
Then I used R to visualize the relationship between contig lengths and the contaminated lengths. There were 14846 contigs, among which 12004 (80.86%) contigs, 2788 (18.78%) contigs and 54 (0.36%) contigs has a contaminated rate of blow 1%, between 1% and 10%, and more than 10% respectively.
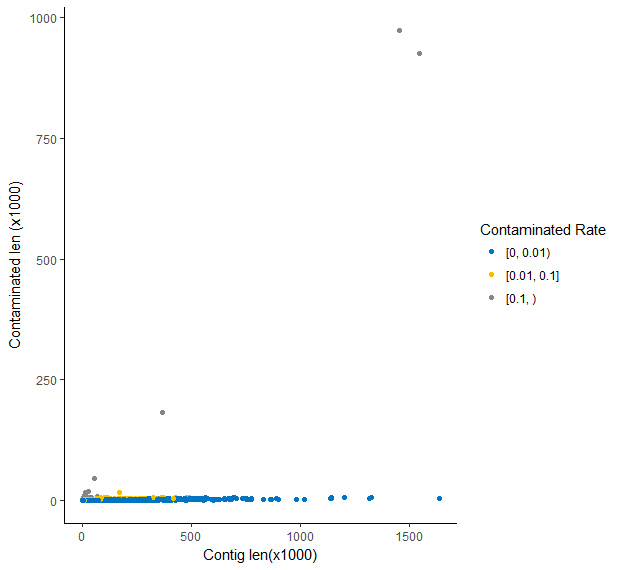
Masked library
1 | classify |
And about 17% of contigs were classified as ‘contaminant’.
1 | head -5 flye.run2.report |
There were 14846 contigs, among which 14766 (99.46%) contigs, 61 (0.41%) contigs and 19 (0.13%) contigs has a contaminated rate of blow 1%, between 1% and 10%, and more than 10% respectively.
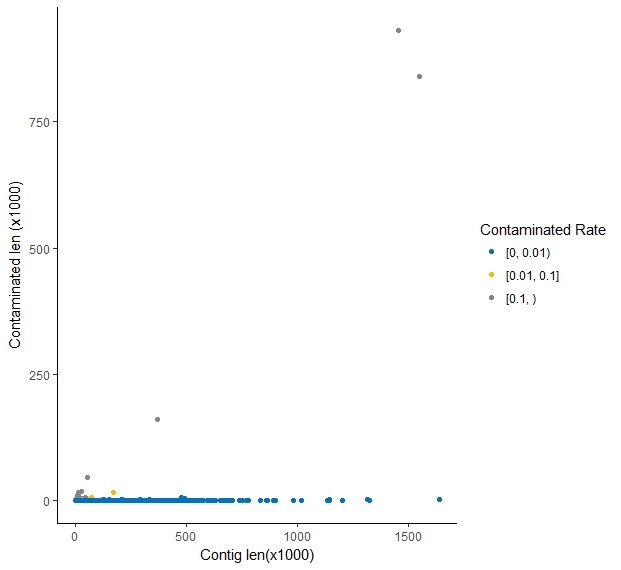
Replace contaminant regions with N
My collaborator asked me to give him an assembly draft for meta-genomic study, which a ‘clean’ one was needed. Removing all the contaminated sequences was not feasible, since there would be few contigs left.
Then I checked the output of Kraken, which contained the detailed information of each contig:
1 | head -3 flye.run2.kraken |
Because Kraken uses a k-mer mapping strategy to locate potential contaminants, there would be many contigs within which only a small part could be aligned to bacteria/virul sequences. So I retrieved the precise k-mers that mapped to contamination, and masked them with ‘N’. I discussed this with another guy: Questions about de novo genome assembly from mixed DNA samples, and he also thought it’s a vivid approach.
Finally
But I began to think how the contaminanted sequences would affect the assembly process, and maybe removing contaminants from raw data is a good way. See discussions here: Filtering for contamination when assembling a genome, before or after assemby? and Question: How to remove contamination from the transcriptome assembly.
Useful links
- refseq2kraken: download refseq-genomic data and prepare it for Kraken
- Download refseq-genomic data and prepare it for Kraken
- Building a low-complexity masked database with dustmasker
- Questions about de novo genome assembly from mixed DNA samples
- Handling microbial contamination in NGS data
- Question: Contamination in assembly
- Filtering for contamination when assembling a genome, before or after assemby?
- Question: How to remove contamination from the transcriptome assembly
Change log
- 20180424: create the note.
- 20180620: add the “refseq2kraken” part.
- 20180808: add “Mask low-complexity regions” and respective parts.