This note is to help me figure out the design schema of annotation packages in Bioconductor. And this note is mainly compiled from:
- Annotation Packages: the big picture. Fantastic slide!
- Introduction To Bioconductor Annotation Packages
- Package ‘AnnotationDbi’ Reference Manual
- Making and Utilizing TxDb Objects
Introduction
Packages in Bioconductor can be divided into three categories: software, annotation and experiment.
All of the ‘.db’ (and most other Bioconductor annotation packages) are updated every 6 months corresponding to each release of Bioconductor. Exceptions are made for packages where the actual resources that the packages are based on have not themselves been updated.
Schema of Annotation Packages
Here is a very representative graph, but not informative-enough.
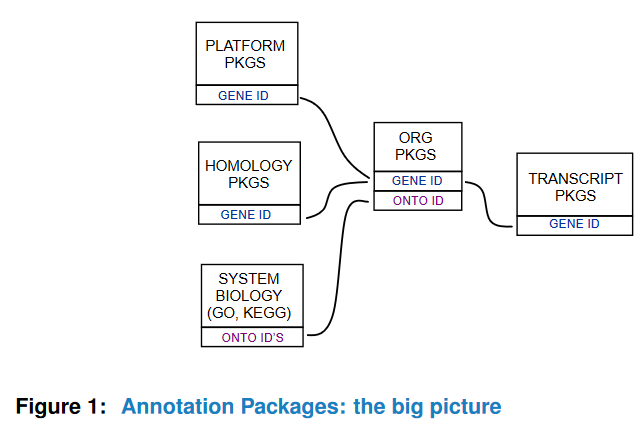
There are three major types of annotation in Bioconductor:
- Gene centric
AnnotationDbpackages:- Organism level: e.g.
org.Mm.eg.db. - Platform level: e.g.
hgu133plus2.db,hgu133plus2.probes,hgu133plus2.cdf. - Homology level: e.g.
hom.Dm.inp.db. - System-biology level: e.g.
GO.db.
- Organism level: e.g.
- Genome centric
GenomicFeaturespackages include:- Transcriptome level: e.g.
TxDb.Hsapiens.UCSC.hg19.knownGene,EnsDb.Hsapiens.v75. - Generic genome feature: can generate via
GenomicFeatures.
- Transcriptome level: e.g.
- One web-based resource accesss
biomart, viabiomaRtpackage:- Query web-based ‘biomart’ resource for genes, sequence, SNPs, and etc.
Working with AnotationDb objects
AnnotationDb is the virtual base class for all annotation packages. It contain a database connection and is meant to be the parent for a set of classes in the Bioconductor annotation packages. These classes will provide a means of dispatch for a widely available set of select methods and thus allow the easy extraction of data from the annotation packages.
All the annotation packages that base on AnnotationDb object expose an object named exactly the same as the package itself.
1 | library(org.Hs.eg.db) |
1 | [1] "OrgDb" |
The more specific classes (the ones that you will actually see in the wild) have names like OrgDb, ChipDb or TxDb objects. These names correspond to the kind of package (and underlying schema) being represented.
Like:
org.Hs.eg.db- Genome wide annotation for Human.TxDb.Hsapiens.UCSC.hg19.knownGene- Annotation package for TxDb object(s).hgu95av2.db- annotations for the hgu95av2 Affymetrix platform.
Methods
select, columns and keys are used together to extract data from an AnnotationDb object (or any object derived from the parent class). Examples of classes derived from the AnnotationDb object include (but are not limited to): ChipDb, OrgDb, GODb, InparanoidDb and ReactomeDb.
columnsshows which kinds of data can be returned for the AnnotationDb object.keytypesallows the user to discover which keytypes can be passed in to select or keys and the keytype argument.keysreturns keys for the database contained in theAnnotationDbobject . This method is already documented in the keys manual page but is mentioned again here because it’s usage with select is so intimate. By default it will return the primary keys for the database, but if used with thekeytypeargument, it will return the keys from thatkeytype.selectwill retrieve the data as adata.framebased on parameters for selected keyscolumnsandkeytypearguments. Users should be warned that if you callselectand request columns that have multiple matches for your keys, select will return adata.framewith one row for each possible match. This has the effect that if you request multiple columns and some of them have a many to one relationship to the keys, things will continue to multiply accordingly. So it’s not a good idea to request a large number of columns unless you know that what you are asking for should have a one to one relationship with the initial set of keys. In general, if you need to retrieve a column (like GO) that has a many to one relationship to the original keys, it is most useful to extract that separately.mapIdsgets the mapped ids (column) for a set of keys that are of a particular keytype. Usually returned as a named character vector, a list or even a SimpleCharacterList.saveDbwill take anAnnotationDbobject and save the database to the file specified by the path passed in to the file argument.loadDbtakes a .sqlite database file as an argument and uses data in the metadata table of that file to return an AnnotationDb style object of the appropriate type.speciesshows the genus and species label currently attached to the AnnotationDb objects database.dbfilegets the database file associated with an object.dbconngets the datebase connection associated with an object.taxonomyIdgets the taxonomy ID associated with an object (if available).
ChipDb
Platfom-based or chip-based annotation package are an extremely common kind of Annotation package. The following examples show how to use standard methods to interact with an object of this type.
1 | library("hgu95av2.db") |
Things loaded along with this package
1 | ls("package:hgu95av2.db") |
1 | [1] "hgu95av2" "hgu95av2.db" "hgu95av2_dbconn" "hgu95av2_dbfile" "hgu95av2_dbInfo" |
These packages appear to contain a lot of data but it is an illusion.
1 | > library(hgu95av2.db) |
Use columns() to see possible values for columns.
1 | columns(hgu95av2.db) |
1 | [1] "ACCNUM" "ALIAS" "ENSEMBL" "ENSEMBLPROT" "ENSEMBLTRANS" "ENTREZID" "ENZYME" "EVIDENCE" |
Use help("xxx")to see the description of columns.
1 | help('SYMBOL') |
Use keytypes() to see possible values for keytypes. In reality, some kinds of values make poor keys and so this list is shorter than that of above.
1 | keytypes(hgu95a.db) |
1 | [1] "ACCNUM" "ALIAS" "ENSEMBL" "ENSEMBLPROT" "ENSEMBLTRANS" "ENTREZID" "ENZYME" "EVIDENCE" |
Use keys() to extract some sample keys back. (default if the primary key.)
1 | head(keys(hgu95av2.db)) |
1 | [1] "1000_at" "1001_at" "1002_f_at" "1003_s_at" "1004_at" "1005_at" |
Or for a particular keytype.
1 | head(keys(hgu95av2.db, keytype='SYMBOL')) |
1 | [1] "A1BG" "A2M" "A2MP1" "NAT1" "NAT2" "NATP" |
Use select() to retrieve data.
1 | #1st get some example keys |
1 | 'select()' returned 1:1 mapping between keys and columns |
If one wants to get only one column of data, mapIds can be used.
1 | mapIds(hgu95av2.db, keys=k, column=c("GENENAME"), keytype="PROBEID") |
1 | 'select()' returned 1:1 mapping between keys and columns |
OrgDb
An organism level package (an ‘org’ package) uses a central gene identifier (e.g. Entrez Gene id) and contains mappings between this identifier and other kinds of identifiers (e.g. GenBank or Uniprot accession number, RefSeq id, etc.).
The name of an org package is always of the form org.<Ab>.<id>.db (e.g. org.Sc.sgd.db) where <Ab> is a 2-letter abbreviation of the organism (e.g. Sc for Saccharomyces cerevisiae) and <id> is an abbreviation (in lower-case) describing the type of cen- tral identifier (e.g. sgd for gene identifiers assigned by the Saccharomyces Genome Database, or eg for Entrez Gene ids).
Using OrgDb packages is just like using ChipDb packages.
1 | library(org.Hs.eg.db) |
GO.db
1 | library(GO.db) |
TxDb
A TxDb package connects a set of genomic coordinates to various transcript oriented features. The package can also contain identifiers to features such as genes and transcripts, and the internal schema describes the relationships between these different elements.
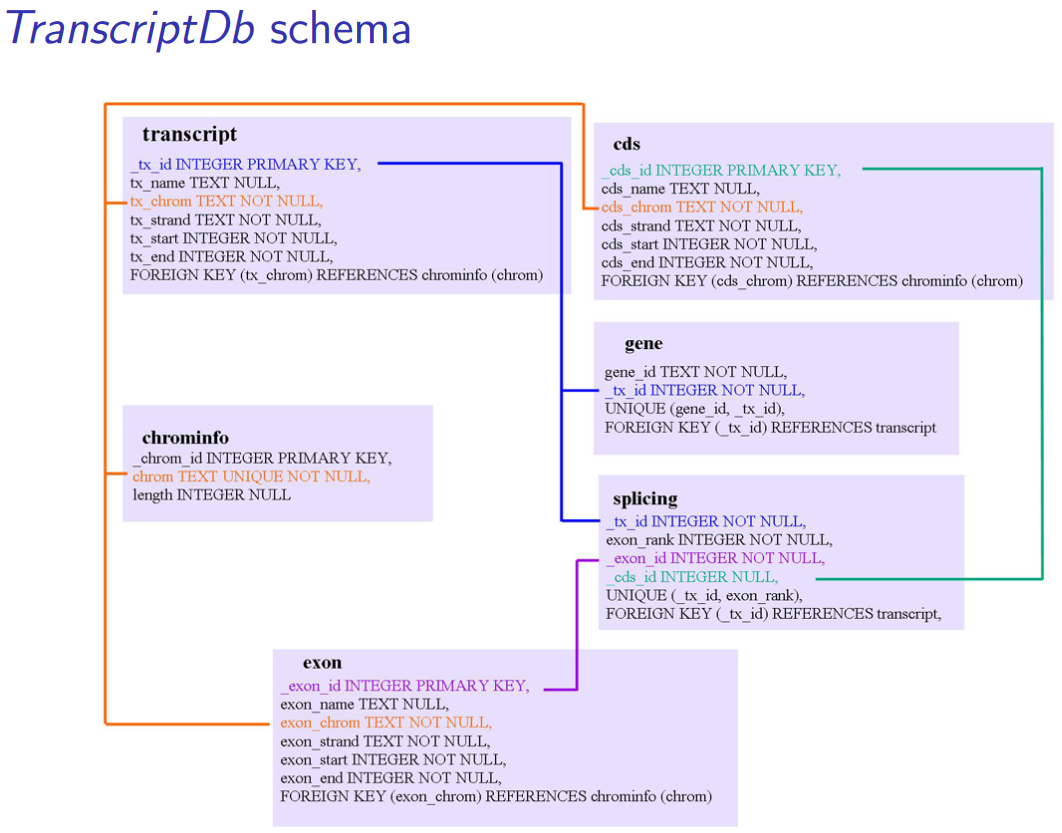
This class maps the 5’ and 3’ untranslated regions (UTRs), protein coding sequences (CDSs) and exons for a set of mRNA transcripts to their associated genome. TxDb objects have numerous accessors functions to allow such features to be retrieved individually or grouped together in a way that reflects the underlying biology.
All TxDb containing packages follow a specific naming scheme that tells where the data came from as well as which build of the genome it comes from.
Package GenomicFeatures contain a set of tools and methods to make and manipulate transcript centric annotation.
1 | library(TxDb.Hsapiens.UCSC.hg19.knownGene) |
1 | TxDb object: |
In addition to accessors via select, TxDb objects also provide access via the more familiar transcripts, exons, cds, transcriptsBy, exonsBy and cdsBy methods, and they will return GRanges objects.
The ‘ungrouped’ functions transcripts, exons, cds, genes and promoters return the coordinate information as a GRanges object.
1 | GR = transcripts(txdb) |
The ‘grouped’ function transcriptsBy, exonsBy, cdsBy, intronsByTranscript, fiveUTRsByTranscript and threeUTRsByTranscript extract genomic features of a given type grouped based on another type of genomic feature.
1 | > GRList <- transcriptsBy(txdb, by = "gene") |
The transcriptsBy function returns a GRangesList class object. The show method for a GRangesList object will display as a list of GRanges objects. And, at the bottom the seqinfo will be displayed once for the entire list.
Then standard GRanges and GRangesList accessors can be used to deal with the returnings. And one can also leverage many nice IRanges methods.
EnsDb
Similar to the TxDb objects/packages, EnsDb objects/packages provide genomic coordinates of gene models along with additional annotations (e.g. gene names, biotypes etc) but are tailored to annotations provided by Ensembl.
The central methods implemented for EnsDb objects allow also the use of the EnsDb specific filtering framework to retrieve only selected information from the database.
1 | library(EnsDb.Hsapiens.v86) |
key() function has an additional filter parameter, which accepts AnnotationFilter object.
1 | keys <- head(keys(edb, keytype="GENEID")) |
1 | [1] "ENSG00000129816" "ENSG00000129845" "ENSG00000131538" "ENSG00000147753" "ENSG00000147761" "ENSG00000176728" "ENSG00000180910" |
keys in mapIds and select also accepts AnnotationFilter object.
1 | txs <- select(edb, keys=list(GeneBiotypeFilter("lincRNA"), SeqNameFilter("Y")), columns=c("TXID", "TXSEQSTART", "TXBIOTYPE")) |
Other Questions
Question: Difference between GO.db, biomaRt, and org.Hs.eg.db in GO annotations
GO.dbandorg.Hs.eg.dbare copies of the GO annotations.GO.dbis updated every 6 months with each release of Bioconductor.org.Hs.eg.dbis also updated at the same time and usingGO.db.biomaRtconnects to the server where the informations is stored, so it will be the most up to date. If you want a stable release you can use eitherGO.dbororg.Hs.eg.db, if you want the most up to date (from yesterday) data every time you do an analysis you can usebiomaRt.
Question: org.Hs.eg.db - hg38 build?
The orgDb packages don’t really contain any positional annotation. They used to, but these days you will be directed to a TxDb package if you try to get positional info. And the TxDb have the build in the package name. The orgDb packages mostly contain mappings between various databases and some functional annotation, none of which is based on any build. In fact, most of that stuff is updated weekly or monthly, so the orgDb packages get outdated to a certain extent rather quickly.
Change log
- 20180918: create the note.